Figure 1: Generating Machine
| [Bioinformatics] and [DNA Compression] |
|
|
We describe a model for strings of characters that is loosely based on the Lempel Ziv model with the addition that a repeated substring can be an approximate match to the original substring; this is close to the situation of DNA, for example. Typically there are many explanations for a given string under the model, some optimal and many suboptimal. Rather than commit to one optimal explanation, we sum the probabilities over all explanations under the model because this gives the probability of the data under the model. The model has a small number of parameters and these can be estimated from the given string by an expectation-maximization (EM) algorithm. Each iteration of the EM algorithm takes O(n2) time and a few iterations are typically sufficient. O(n2) complexity is impractical for strings of more than a few tens of thousands of characters and a faster approximation algorithm is also given. The model is further extended to include approximate reverse complementary repeats when analyzing DNA strings. Tests include the recovery of parameter estimates from known sources and applications to real DNA strings.
Partly
supported by the Australian Research Council,
ARC grant A49800558
Copyright © 1998, American Association for Artificial Intelligence
[www.aaai.org].
All rights reserved.
pattern discovery, repeats, sequence analysis, hidden Markov model, DNA, data compression.
We describe a model for strings of characters which is loosely based on the Lempel Ziv (1976) model that a string consists of a mixture of ``random'' characters and repeated substrings. The new model has the addition that a repeated substring need not match the original substring exactly but may match it approximately due to the presence of variations, e.g. mutations, experimental error and noise. When modelling DNA sequences, we also allow approximate reverse complementary repeats. The model has a small number of parameters governing the probability of repeats, the probability distribution of the lengths of repeats and the probability of differences within repeats. The parameters can be estimated from a given string by an expectation maximization (EM) algorithm (Baum and Eagon 1967) (Baum et al 1970) (Dempster, Laird, and Rubin 1977). Each iteration of the EM algorithm takes O(n2) time and a few iterations are generally sufficient. An approximation algorithm that runs in near linear time is available for long strings where O(n2) complexity is too great.
The primary purpose of the work is not to compress strings, and in particular DNA strings, so as to save computer storage space or to reduce data transmission costs. Rather, the purpose is to model the statistical properties of the data as accurately as possible and to find patterns and structure within them. As such our algorithms do not have to run as quickly as typical file compression programs although they could act as benchmarks for such programs in this application area. In fact the final compression step is not carried out, instead probabilities and the lengths of encodings are calculated although actual encodings could be produced in principle (Wallace and Freeman 1987).
Agarwal and States (1994), Grumbach and Tahi (1994), and others have recognised the general importance of compression for pattern discovery in biological (and other) sequences. One of the benefits of looking at pattern in this way is a hypothesis test: the claimed discovery of pattern, structure or repetition is only an ``acceptable hypothesis'' if it leads to genuine compression of the data. Using compression as the criterion for what is variously known as inductive inference or machine learning dates back at least to the work of Solomonoff (1964), Kolmogorov (1965), Chaitin (1966) and Wallace and Boulton (1968). It is widely held that the crucial part of compression is accurate modelling of the data and that the degree of compression indicates the accuracy of the models used. A practical advantage of thinking about inductive inference in compression terms is that it becomes obvious that all relevant information, and in particular any inferred parameters of a model, other than common knowledge, must be included if the encoded data is to be decodable, i.e. comprehensible. The large body of standard methods, algorithms and heuristics from data compression can also be used. Lastly, the scientific method expects theories and hypotheses to make testable predictions, and only good predictions can be used to produce brief encodings.
Interestingly, common file compression programs perform poorly on DNA sequences; something more specialised is needed and any available prior knowledge or expertise in the biological domain should be brought to bear. Milosavljevic and Jurka (1993) applied a Lempel Ziv type model to DNA and Powell et al (1998) examined the compression available in variations on that work. Wootton (1997) directed attention to what was termed the ``compositional complexity'' of strings, i.e. the multistate distribution (Boulton and Wallace 1969) of characters in sliding windows, and stopped short of providing a figure for the overall information content of a complete string. Loewenstern and Yianilos (1997) extended a file compression algorithm to DNA by allowing mismatches to occur in ``contexts'' with considerable improvement over previous methods; the model has several dozen parameters which are fitted to the data but do not have direct biological interpretations. Rivals and Duchet (1997) presented a heuristic which can join exact repeats that are close together into clusters or runs, i.e. it can make some allowance for approximate repeats.
In the following sections we recall the Lempel Ziv model, extend it to allow approximate repeats, and present an inference algorithm and tests on known sources. A faster, approximate algorithm is described for longer strings.
The basic Lempel Ziv model (1976) considers a string to be made up
of a mixture of random characters and repeated substrings.
For example, one possible explanation of the string
AAGTACACGTACAGT
under the model is AAGTACAC(3,5)GT
where (3,5) indicates a repeat from position 3 of length 5,
i.e. GTACA.
Random characters are drawn from an alphabet with
some probability distribution,
in the simplest case a uniform distribution.
A repeated substring is a copy of a substring that starts
somewhere earlier in the string.
The statistical properties of repeats are
the probability with which repeats occur,
the probability distribution on starting points and
the probability distribution on the lengths of repeats.
Many variations on the Lempel Ziv model are possible but simple choices are for a fixed probability of repeats and a uniform distribution (from 1 to the current position-1) on starting points. Ziv and Lempel (1977) demonstrate that their model asymptotically converges, in terms of compression, to the true model when the data come from some fixed but unknown source. This implies that theirs is a good model to use when the true nature of the source is unknown.
To compress a string under the Lempel Ziv model,
one outputs code words describing either a character or a repeat.
Assuming a fixed probability of a repeat, P(start),
and a uniform probability distribution over characters,
a random character has a code word of length
-log2(1-P(start))+log2(|alphabet|)
bits.
Assuming a uniform distribution on the source of repeats,
a repeat into position i has a code word of length
-log2(P(start))+log2(i-1)+log2(P(length))
bits.
The usual objective is to find a single optimal explanation
for the given string, using this as the basis for compressing it.
Finding an optimal explanation is the search problem.
We know that in the related sequence alignment problem
(Allison, Wallace, and Yee 1992),
a single optimal explanation underestimates the probability of the data
and gives biased estimates of parameter values.
Therefore we sum over all explanations under the new model which gives better
compression, particularly for ``weak'' or ``statistical'' structure.
The Lempel Ziv model inspired many file compression programs. To be practical, such programs must run in linear time, and in fact linear time with a small constant, so much of the work in file compression has been on sophisticated algorithms and data structures, such as hash-tables and suffix-trees, to achieve this end. The current practice in general text compression is to keep a data structure of ``contexts'' i.e. words of some length k, that have occurred in the past, with statistics on the frequencies of characters that followed instances of each context. Major variations cover whether or not there is a bound on context length (Cleary and Teahan 1997), and how to balance predictions from long but rare contexts against those from short but more frequent contexts.
When typical file compression programs, such as zip, are run on DNA strings they often fail to compress the DNA below the base-line figure of two bits per character (Loewenstern and Yianilos 1997), i.e. they fail to compress the DNA at all. This is due to a combination of factors: File compression programs are generally set to deal with an alphabet of size 256, perhaps with some expectation of the Ascii character set (about 100). Most DNA seems to have rather little redundancy, with compression to 1.9 or 1.8 bits per character being possible with specialist programs. File compression programs do gradually adapt to the DNA's statistics but have often lost too much ground in doing so to make it up on sequences such as HUMHBB (73 thousand characters).
The current work uses a new model of strings where a repeated substring can be an approximate, rather than an exact, match to the original substring. There are two prior reasons to believe that this could be useful. First, it in effect increases the number of contexts that match the last characters of the string, at least approximately if not exactly, and therefore increases the amount of available information about what could come next. Second, for some data sets and notably for DNA there are well known processes by which instances of repeats can differ from each other.
Events in the replication of DNA strings can lead to the duplication of substrings (repeats) and also duplication from the complementary strand in the reverse direction (reverse complementary repeats). These events can create repeats with lengths of hundreds or thousands of characters. Once a substring has been duplicated, the individual copies are subject to the usual evolutionary processes of mutation by which characters can be changed, inserted and deleted. The subsequent mutation process is well modelled by variations on the edit-distance problem which also model spelling errors in text and errors and noise in many other kinds of sequence.
Some substrings occur a great many times in DNA.
For example, the Alu sequences (Bains 1986),
of length about 300 characters,
appear hundreds of thousands of times in Human DNA
with about 87% homology to a consensus Alu string.
Some short substrings such as TATA-boxes,
poly-A and (TG)*
also appear more often than by chance.
Methods are known
(Allison and Yee 1990) (Allison, Wallace, and Yee 1992)
for calculating the probability
of two DNA strings, S1 and S2, given the hypothesis
that S1 and S2 are related,
P(S1&S2|related),
for some simple hidden Markov models of mutation.
The complementary hypothesis is that S1 and S2 are unrelated
and gives a -log2 probability of about two bits per character
for random strings,
P(S1&S2|unrelated)=P(S1).P(S2),
i.e.
-log2(P(S1&S2|unrelated)) =
-log2(P(S1))-log2(P(S2)).
If S1 and S2 are substrings of some larger string,
and it is assumed that S2 is an approximate repeat of S1,
we get
P(S1&S2|related) = P(S1).P(S2|S1&related)
and the -log2 of the last part,
-log2(P(S2|S1&related)),
is the cost of encoding the changes in S1's approximate copy, S2.
This encoding is compact if S1 and S2 are similar.
An alignment of S1 with S2 gives a way of editing (mutating) S1 into S2. An encoding of these edit operations can be taken as an upper bound of the -log2 probability of S2 given the related S1, e.g.
Alignment:
S1: ACGTAC-T
| || | |
S2: A-GTTCGT
Edit S1 -> S2:
copy, delete, copy, copy,
change(T), copy, insert(G), copy
Typically there are a great many ways of editing S1 into S2, some optimal and some sub-optimal; these are mutually exclusive hypotheses and together exhaust all the ways in which S1 and S2 can be related under the model. If there are two optimal explanations, adding their probabilities saves one bit in compression. Each suboptimal explanation has a lower probability but there are a great many more of them. For example, if there are two options for a plausible alignment early in the data shortly followed by two more options, these options multiply together to give four options in total - and this is just the start. Adding the probabilities of all such editing sequences yields the probability, under the model, of S2 given S1 and given S2 being related to S1. This calculation can still be carried out in O(n2) time by a modified dynamic programming algorithm (DPA) that sums probabilities (Allison, Wallace, and Yee 1992) rather than maximizing the probability of an alignment (edit sequence) which is equivalent to minimizing the edit-distance. We therefore use this alignment algorithm, summing the probabilities of all alignments of possible repeats, within the string compression model. The algorithm actually works with the -log2 of probabilities but it is sometimes convenient to discuss it in terms of the probabilities directly.
|
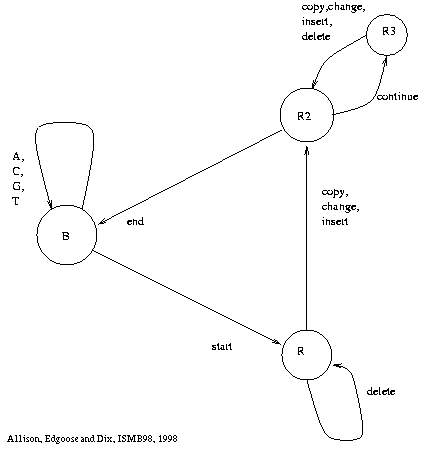
Figure 1: Generating Machine |
Figure 1 shows a finite state machine
for generating strings under
the new model;
it is a mathematical abstraction derived from the considerations above.
From the base state, B,
the machine can generate ``random'' characters, returning to the base state.
It can also start a repeat, moving to state R.
From state R, characters can be copied from the source substring,
but characters can also be changed, inserted or deleted.
The auxiliary states, R2 and R3, are simply there to ensure
that invisible events are prohibited,
i.e. at least one character must be output before returning to B.
The repeat ends with a return to the base state.
The base state is also the start and end state of the machine.
Many variations on the ``architecture'' of the machine are possible
to incorporate prior knowledge while staying within the general
framework.
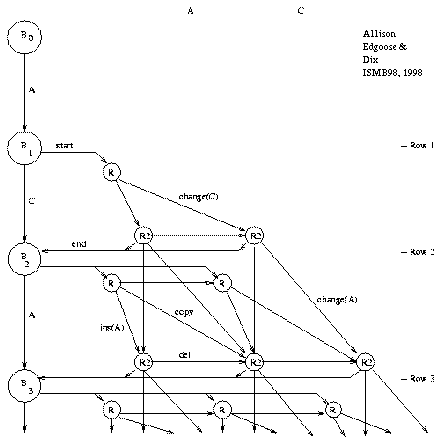 Figure 2: Repeat Graph |
Figure 2 shows the ``repeat graph'' for a string
beginning ACA... under the new model;
note that the graph is acyclic.
A node represents a machine state at a particular time.
The graph amounts to an unfolding of all possible sequences
of state transitions of the machine described above (figure 1),
except that state R3 is merged into R2 for simplicity.
A path through the graph from the start node, B_0, to the end node, B_n, is an ``explanation'' of the string. The paths form an exclusive and exhaustive set of explanations under the model. Some explanations are plausible and some are quite implausible, but all of their probabilities are summed by the DPA. The left-hand column of the repeat graph contains state-transitions of the base state that generate ``random'' characters. A repeat may start from any point in this column. Once within a repeat, the repeat may end or continue. If the repeat continues, a character is copied or changed (diagonal move), deleted (horizontal move) or inserted (vertical move). The repeat's origin must be encoded when it starts. A new character must be encoded with each insert or change but this is not necessary for a copy or delete. The length of a repeat is effectively encoded in a unary code, by repeatedly stating that the repeat has not yet ended. A unary code corresponds to a geometric probability distribution. It is not claimed that a geometric distribution is an optimal fit to the distribution of repeat lengths; it is used because it has the property of making the DPA's incremental calculations ``local'' which permits the summing of all explanation probabilities in O(n2) time. This is discussed in a later section.
Reverse complementary repeats were left out of the discussion above but are included in the model by a further set of states R', R2' and R3' in the machine, and nodes and arcs in the repeat graph, similar to those for forward repeats in figure 1 and figure 2. There is a corresponding set of probability parameters for the start, continuation and mutation of reverse complementary repeats.
The various probability parameters can be given apriori if they are known. Alternately they can be (re)estimated in an expectation maximization (EM) process: Each node in the repeat graph (figure 2) contains the average frequencies of transition types over all paths leading to the node from the start node. When two paths meet, a weighted average of their counts is formed, weighted by the paths' relative probabilities. The frequencies in the end-node are used to derive new probability estimates for the next EM iteration. The EM process is guaranteed to converge because the new parameter values must be as good or better than the old values for the current path weightings and, similarly, the next set of path weightings will be as good or better than the old ones for the new parameter values. Convergence to a local optimum is possible in principle but this is not a problem in practice. The process terminates when the overall probability increase for an iteration is less than some small limit. A few iterations are sufficient if the initial values are ``sensible''. The statement of the parameter values, to optimum accuracy, must be included in the final -log probability calculations for the string if one is to legitimately compare simple and complex models; this is done using the calculations of Boulton and Wallace (1969) for multistate distributions.
A further improvement is to use a first-order Markov model in place of the random-character (uniform, zero-order Markov model) part of the model for the base-state transitions as this is found to give an improvement of 0.03 bits per character on HUMHBB, for example. In addition, characters involved in changes are assumed to come from the underlying probability distribution for the alphabet renormalised after the removal of the character being changed.
There are clearly O(n2) nodes in the repeat graph
but the algorithm only needs to keep two rows,
the previous row and the current row, to operate in O(n) space.
The algorithm takes O(n2) time per EM step.
Importantly, the algorithm fails to compress truly random, artificial DNA strings. The inferred value of P(start) is close to zero for such data but nevertheless the string requires more than two bits per character and the hypothesis that it contains any pattern or structure fails the significance test.
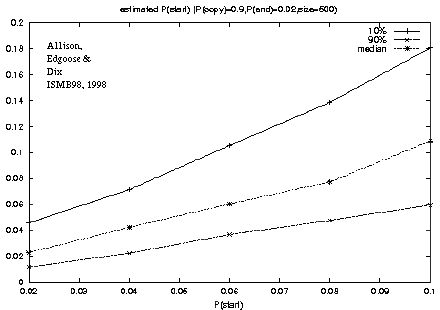 Figure 3: Recovery of the P(start) model parameter from a known source. |
The ability of the algorithm
to recover the parameter values of a known source was tested:
A program was written to generate random strings according to the new model
with specified parameter values.
One parameter was varied systematically while the others were held constant.
For each parameter setting, 100 strings of length 500 were generated.
For example, figure 3
shows a plot of the genuine versus estimated values of P(start)
for repeats.
Although the generating parameters were varied systematically
there is naturally variation in transition frequencies in particular strings,
as indicated by the gap between the 10% and 90% percentile estimates.
The median shows good recovery of the generating value,
for repeats with P(start) in the range tested, between 0.02 and 0.1.
For real DNA strings P(start) is likely to be under 0.1.
Similar tests show good recovery of the other parameters of
the model.
The O(n2) algorithm previously described takes thirty minutes per iteration on a Silicon Graphics ``Indy'' work-station for Dromaster (six thousand characters) and two days per iteration for human globin region HUMHBB (73 thousand characters). A faster approximation algorithm is available if greater speed is necessary. This operates by summing the probabilities for only those explanations in the most ``important'' areas of the repeat graph (figure 2) to give an upper bound on the string's true -log2 probability.
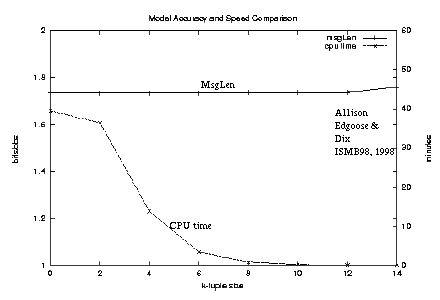 Figure 4: Comparison of model accuracy and speed |
The idea is to find (most) good explanations and only run the DPA in regions
close (+/-5 nodes) to a good explanation.
A hash-table is used to record all instances of each k-tuple in the string
where k is a constant, typically in the range 6 to 14.
Most significant repeats are likely to contain exact matches of length k,
from time to time.
A region is ``turned on'' at a k-tuple match.
A region is left turned on so long as any of its nodes is contributing
a not insignificant amount to the probability of the string,
currently assessed as having a -log2 probability of no more than
that of the base-state minus the repeat start-up cost plus 4 bits.
A region can grow or shrink and eventually be turned off
depending on the contributions within it.
This approximation gives a trade-off between accuracy and speed.
Making the regions wider,
the value of k smaller,
and the threshold on probability contributions more lenient
all increase accuracy at the price of speed.
Figure 4
plots the relationship between calculated
-log2 probability and running-time
for the approximation algorithm as k is varied,
the string being 6000 characters from position 23,000 of HUMHBB
which contains one significant repeat.
It is quite possible that continued tinkering with this or different heuristics
may lead to a better trade-off between accuracy and
speed.
|
The approximation algorithm and, where practical, the exact algorithm were run on real DNA strings. Dromaster is a gene, from drosophila melanogaster, for a repetitive protein so the cDNA is also quite repetitive. At 6.3K, the O(n2) algorithm can comfortably be run on it. HUMHBB is the human globin region and contains multiple globin genes. Tobacco chloroplast, CHNTXX, is notable for a 25K reverse complementary repeat. Yeast chromosome III was included as a long string, 315K nucleotides.
Table 1 gives the coding figures for
biocompress2 (Grumbach and Taji 1994) and
CDNA-compress (Loewenstern and Yianilos 1997),
the first two columns coming from the latter paper,
compared with the new string model.
The figures for HUMHBB, CHNTXX and Yeast chrIII
are from the approximate algorithm set to run in realistic times
and are thus upper-bounds on the
true entropies under the model.
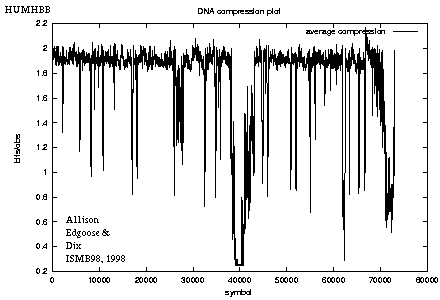 Figure 5: Compression plot for HUMHBB |
Figure 5 shows the code length per character as a moving average over 100 positions for HUMHBB. For example, the drop around position 40,000 corresponds to a strong repeat from the gamma-globin genes, hbgg and hbga.
Examining the inferred probability parameters tells us something
general about a string.
Dromaster has numerous forward repeats (P(start)=0.015)
that are short (P(end)=0.06)
and similar (P(copy)=0.94).
It has no reverse complementary repeats (P(start)=0.0001).
CHNTXX has a few reverse complementary repeats (P(start)=0.00005)
that are long (P(end)=0.0002)
and high-fidelity (P(copy)=0.9999);
i.e. it is dominated by the 25K example.
The
length of a repeat in the new model is effectively encoded by a unary code
which corresponds to a geometric probability distribution,
as mentioned previously.
Ignoring the question of substring differences,
the code word for a repeat of a given length>0 requires
-log2(P(start))
+ log2(target-1))
- (length-1).log2(P(continue))
- log2(P(end)) bits.
This is a linear function with a slope of
-log2(P(continue)).
It is not claimed that a geometric distribution is the perfect model
for the lengths of repeats, indeed that is most unlikely,
but it does make the DPA's calculations in the repeat graph ``local''
because the incremental cost of extending all repeats is the same.
This allows an O(n2) DPA to sum the probabilities of all
paths through the graph (figure 2).
A mixture of two (or more) geometric distributions
has the same property although at the price of roughly doubling (etc.)
the number of states in the graph.
In passing, note that it would be possible to change to
arbitrary probability distributions having concave negative logs
by adapting the technique of Miller and Myers (1988),
provided that one wanted only a single optimal explanation of the string.
The resulting algorithm would have O(n2) complexity when a certain equation
is solvable in O(1) time, and O(n2 log(n)) complexity otherwise.
A model with a mixture of two types of forward repeats and two types of reverse complementary repeats was run on HUMHBB: It marginally reduced the entropy from 1.728 to 1.725 bits/character, covering the cost of stating the extra sets of parameters so this is not a case of over-fitting. The parameter estimates describe mixtures of repeats: Low fidelity (P(copy)=0.78) forward repeats occur most often (P(start)=0.0006). High fidelity (P(copy)=0.96) forward repeats occur less often and are shortest on average (P(end)=0.006). Low fidelity (P(copy)=0.70) reverse complementary repeats occur rarely (P(start)=0.0002). Medium fidelity (P(copy)=0.86) reverse complementary repeats occur rarely but are longest on average (P(end)=0.003). Over all types of repeat, changes are about ten times more probable than inserts or deletes.
In a similar vein to the repeat-length distribution, the present algorithms use the simplest possible model of point-mutations on repeated substrings. As above, this effectively codes the length of a run of inserts or deletes with a unary code. This is not unreasonable because inserts and deletes do seem to occur at fairly low rates in repeats. However, one could use linear gap-costs (Gotoh 1982), and even piecewise-linear gap-costs, while still summing over all explanations as described for aligning two strings (Allison, Wallace, and Yee 1992). That technique could be used in the current string model at the cost of increasing the number of states in the repeat graph, leaving the exact algorithm's complexity at O(n2) but with a larger constant.
Note that the new model and its algorithms have an association with Tichy's (1984) block-moves model of string comparison. The latter involves a ``source'' string and a ``target'' string, that is to say it is not symmetrical with respect to S1 and S2. A substring, i.e. a block, can be copied from the source string and appended to the target string as the latter is being built up; in fact this is the only way in which the target can grow. Tichy's objective was to find the explanation of the target string that requires the smallest number of these block-moves. This would be a most probable explanation provided that all block-moves were equally probable. The copies are exact and there is a linear-time algorithm for the problem. In a parallel, our new string model could be used to compare two strings S1 and S2 under ``approximate block-moves'': append S1 and S2, now compress the result under the new model but only allowing repeats from S1 into S2. In fact it would make sense also to allow repeats from S1 into S1 and from S2 into S2. We speculate that this might be another useful way to look for relationships between biological sequences.
The new model of strings allows approximate repeated substrings; a repeated substring need not be an exact match to the original but may contain changes, insertions and deletions. This allows more (approximate) contexts from the past to play a part in predicting the next character. It also directly models, to some extent, actual events in the replication of DNA. Consequently the method's parameter estimates can be directly related to these events. Tests on known artificial sources show good estimation of parameter values. There is a natural significance test for any structure, pattern or repetition claimed to have been found.
A dynamic programming algorithm calculates the probability of a string under the model in O(n2) time when the probability parameters governing repeats are given in advance. The algorithm can be used as the step in an expectation maximization algorithm to estimate the parameters, if they are not known in advance, and a few iterations are usually sufficient. A faster approximation algorithm is available when greater speed is necessary. The algorithms give good compression of real DNA sequences indicating that the patterns found are significant.
A two-dimensional plot, figure 6,
using intensity to show the level
of contributions to the string's probability
gives a useful visualization of the identities
and significance of repeated substrings, here for HUMHBB.
The inset magnifies the structure of the reverse complementary repeats
near the end of this string.
Such plots resemble familiar dot-matrix plots
but with a formal connection between significance and grey-scale
level.

See [Molecular Biology] and [DNA Compression].