25. Adding reactivity to structure 2:
Oxygen-isotope-exchange rates in three isostructural oxide ions
December 6, 2010

Abstract: To understand how oxide structures react at the molecular scale, rates of steady oxygen-isotope exchanges were followed in three isostructural molecules of ca 40 atoms as a function of solution composition. These molecules were chosen because the structures in solution are known with complete confidence, yet isotope-exchange reactions can be followed spectroscopically at individual oxygens. The series of molecules differ only in a single Ti(IV) --> substitution in one of the three metal sites, making a series of structures having the stoichiometries: [HxNb
Villa, E. M., Ohlin C. A., Casey W. H. Adding reactivity to structure 2: Oxygen-isotope-exchange rates in three isostructural oxide ions American Journal of Science , 2010, 310, 629-644.
Department of Chemistry and Department of Geology, University of California, Davis, CA
24. The first peroxotitanoniobate cluster
- [N(CH3)4]10[Ti12Nb6O38(O2)6]
June 10, 2010
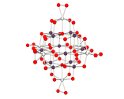
Abstract The first example of a discrete peroxo-polyoxotitanoniobate is introduced and characterized by x-ray crystallography.
Ohlin, C. A.,[1,2] Villa, E. M.,[1,2] Fettinger, J. C.,[1] Casey, William H.[1,2] The first peroxotitanoniobate cluster - [N(CH3)4]10[Ti12Nb6O38(O2)6] Inorg. Chimica Acta, 2010, 363(15), 4405-4407.
1. Department of Chemistry, University of California, Davis, CA.
2. Department of Geology, University of California, Davis, CA.
23. Borate Accelerates Oxygen-Isotope
Exchange for Polyoxoniobate Ions in Water
May 26, 2010
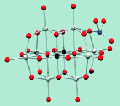
Abstract Understanding simple oxygen exchange reactions is important to a variety of communities concerned with the chemistry of oxides with water. Limitations in the methods available for studying reactions at these oxide-water interfaces, as well as difficulties in characterizing their structures, have led to the use of polyoxometalates (POMs) as model molecules. POMs are metaloxide ions comprised of group 5 and 6 metals. These ions constitute discrete and often soluble clusters than can be spectroscopically probed with great confidence. In addition, POMs are interesting in their own right owing to their structural and chemical diversity, and are finding an increasing number of applications. We have been investigating the oxygen-isotope exchange kinetics in these ions and aqueous solution by 17O Nuclear Magnetic Resonance (NMR) to help better recognize what controls molecule-water interface processes on the level of individual oxygen sites. These structures are chosen because the isotope-exchange reactions could be followed separately from dissociation or condensation of the structure.
Villa, E. M., Ohlin, C. A, Casey, W. H..Borate Accelerates Oxygen-Isotope Exchange for Polyoxoniobate Ions in Water Eur. Chem. J. , 2010, 16(29), 8631-8634.
Department of Chemistry and Department of Geology, University of California, Davis, CA
22. Oxygen-isotope exchange rates for three
isostructural polyoxometalate ions
March 8, 2010

Abstract We compare oxygen-isotope exchange rates for all structural oxygens in three polyoxoniobate ions that differ by systematic metal substitutions of Ti(IV)=>Nb(V). The [HxNb10O28 ](6-x)-, [HxTiNb9O28](7-x)-, and [HxTi2 Nb8O28](8-x)- ions are all isostructural, yet have different Brönsted properties. Rates for sites within a particular molecule in the series differ by at least 104, but the relative reactivities of the oxygen sites rank in nearly the same relative order for all ions in the series. Within a single ion, most structural oxygens exhibit rates of isotopic exchange that vary similarly with pH, indicating that each structure responds as a whole to changes in pH. Across the series of molecules, however, the pH dependencies for isotope exchanges and dissociation are distinctly different, reflecting different contributions from proton- or base-enhanced pathways. The proton-enhanced pathway for isotope exchange dominates at most pH conditions for the [HxTi2 Nb8O28](8-x)- ion, but the base-enhanced pathways are increasingly important for the [HxTiNb9O28](7-x)- and [HxNb10O28](6-x)- structures at higher pH. The local effect of Ti(IV) substitution could be assessed by comparing rates for structurally similar oxygens on each side of the [HxTiNb9O28](7-x)- ion and is surprisingly small. Interestingly, these nanometer-size structures seem to manifest the same general averaged casey +geo amphoteric chemistry that is familiar for other reactions affecting oxides in water, including interface dissolution by proton- and hydroxyl-enhanced pathways.
Villa, E. M., Ohlin, C. A., Casey, W. H. Oxygen-isotope exchange rates for three isostructural polyoxometalate ions J. Am. Chem. Soc., 2010,132(4), 5264-5272.
Department of Chemistry and Department of Geology, University of California, Davis, CA
21. Dissolution of insulating oxide materials at
the molecular scale
March 7, 2010
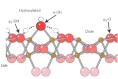
Abstract Our understanding of mineral and glass dissolution has advanced from simple thermodynamic treatments to models that emphasize adsorbate structures. This evolution was driven by the idea that the best understanding is built at the molecular level. Now, it is clear that the molecular questions cannot be answered uniquely with dissolution experiments. At the surface it is unclear which functional groups are present, how they are arranged, and how they interact with each other and with solutes as the key bonds are activated. An alternative approach has developed whereby reactions are studied with nanometre-sized aqueous oxide ions that serve as models for the more complicated oxide interface. For these ions, establishing the structure is not a research problem in itself, and bond ruptures and dissociations can be followed with much confidence. We review the field from bulk -dissolution kinetics to the new isotope-exchange experiments in large oxide ions.
Ohlin, C. André;[1,2] Villa, Eric M.;[1,2] Rustad, James R.,[2] Casey, William H. [1,2] "Dissolution of insulating oxide materials at the molecular scale ", Nature Mat., 2010, 9, 11-19.
1. Department of Chemistry, University of California, Davis, CA.
2. Department of Geology, University of California, Davis, CA.
